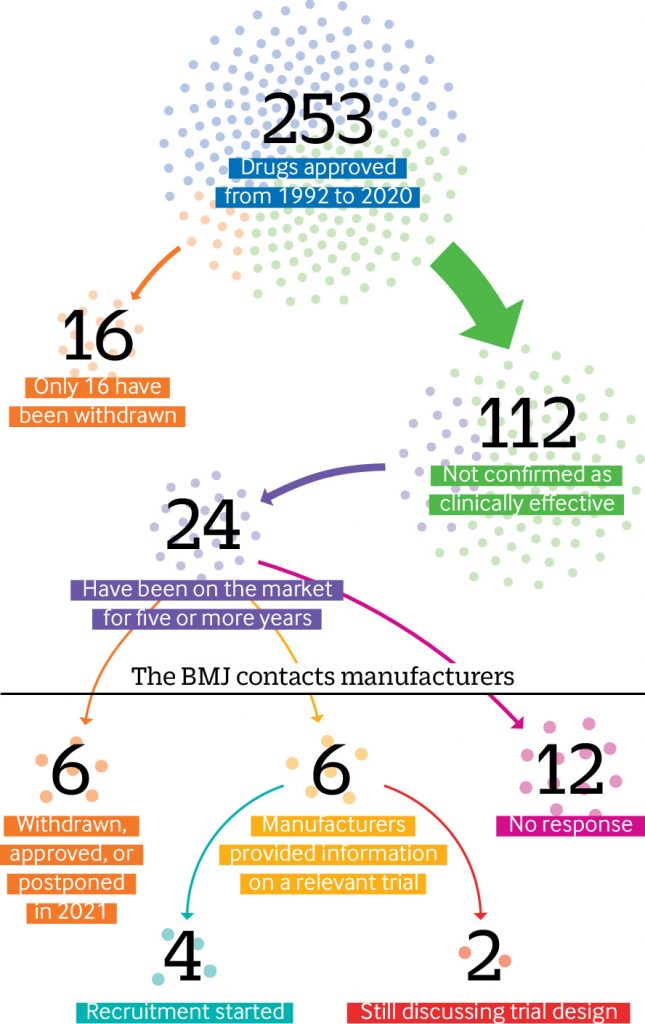
Since the US Food and Drug Administration (FDA) established its accelerated drugs approval pathway in 1992, nearly half (112) of the 253 drugs authorized have not been confirmed as clinically effective, an investigation by The BMJ has found. In a press statement, The BMJ says the “process is plagued by missing efficacy data and questionable evidence.” They also say that experts believe the standards for evidence are too low.
Within the integrative health field, doctors say they are concerned that patients are being prescribed ineffective and/or unnecessary medications, the costs of these medications may be grossly expensive, which places patients at-risk for financial difficulty. They add, FDA has a responsibility to provide greater oversight for all medications, including fast tracked drugs.
“Sixty nine percent of adults aged 40–79 take at least one prescription and twenty-four percent take over five. We find ourselves in an epidemic of overprescribing,” says naturopathic doctor Amy Rothenberg, ND. “While there are many situations when pharmaceuticals are indicated, essential, and effective, clearly there are too many people taking too many drugs. For many ailments, lifestyle and natural medicine approaches work well to support the human body’s capacity for healing and resilience.”
The BMJ Accelerated Drugs Analysis
Elisabeth Mahase, clinical reporter at The BMJ, carried out an in-depth analysis of FDA data from 1992 up to 31 December 2020 and found that of these 112 drugs approved in the last 28 years, 24 (one fifth) have been on the market for more than five years and some have been on the market for more than two decades – often with a hefty price tag.
The accelerated pathway program began to allow drugs into the market before efficacy had been proven. Mahase explains that as part of this approval, the manufacturer must conduct post-approval studies—known as phase IV confirmatory trials—to “verify the anticipated clinical benefit.” If these trials show no benefit, the drug’s approval can be cancelled.
However, according to the analysis done by The BMJ, the data revealed that only 16 drugs approved through this pathway have ever been withdrawn. Most of these were shown to lack efficacy, but in some cases the confirmatory trials were never done.
One example is Celecoxib (Celebrex), which was given accelerated approval in 1999 for the treatment of familial adenomatous polyposis (a genetic disorder that carries a high risk of bowel cancer). It remained on the market for 12 years before the FDA finally asked Pfizer to voluntarily withdraw it for the given indication because the efficacy trials had never been done.
The BMJ asked the manufacturers of 24 treatments that have been on the market for more than five years whether they had conducted phase IV trials. Six drugs had been withdrawn, approved, or postponed.
- Out of the remaining 18 drugs, just one third of manufacturers (6/18) provided information on a relevant trial.
- And of these only four had started to recruit patients, while two companies said they were still in discussion with the FDA over the final study design.
“Despite the pathway’s good intentions to accelerate ‘the availability of drugs that treat serious diseases’ experts are concerned that it is now being exploited – to the detriment of patients, who may be prescribed a drug that offers little benefit and possible harm, and to taxpayers,” writes Mahase.
Huseyin Naci, associate professor of health policy at the London School of Economics, expressed concern, saying “we may have drugs on the market that don’t have any benefits, but certainly predictably have harms associated with them.”
Problem First Reported in 2015
In as far back as 2015, a review of the FDA’s expedited pathways by the US Government Accountability Office said that its “data on post-market safety issues and studies were found to be incomplete, outdated, [and] to contain inaccuracies.” The GAO report cited the following issues:
- “FDA lacks reliable, readily accessible data on tracked safety issues and postmarket studies needed to meet certain postmarket safety reporting responsibilities and to conduct systematic oversight.”:
- “Tracked safety issues are potential safety issues that FDA determines are significant and that it tracks using an internal database.”
- “Internal control standards for federal agencies specify that information should be recorded in a form and within a time frame that enables staff to carry out their responsibilities and that relevant, reliable, and timely information should be available for external reporting purposes. However, evaluations conducted by Center for Drug Evaluation and Research (CDER) of data in its database revealed problems with the completeness, timeliness, and accuracy of the data.”
In April 2021, the Institute for Clinical and Economic Review (ICER) reported that a lack of “credible threats” to withdraw approval if companies didn’t carry out confirmatory trials meant that study sponsors had little incentive to do them.
When efficacy is unclear, the FDA uses indirect (surrogate) measures of clinical benefit. In some cases, there is a strong indication that the surrogate measure predicts meaningful benefit, explains Mahase, but where the situation is less straightforward, “inconsistencies and a lack of transparency surrounding decisions have led to serious questions over the standards of evidence being accepted.”
Rachel Sachs, an associate professor of law at Washington University, said, “There are some instances where the companies really do seem to be taking advantage of the accelerated approval pathway and are using it in a way that makes it harder to get at the truth about whether these products really are safe and effective.”
“It is important that we have regulation and oversight of all agencies that affect the health of Americans. All organizations and regulatory bodies can benefit from a critical analysis of their activities and effectiveness. Having worked on the Washington State Healthcare Technology Assessment Clinical Committee for nine years I am very familiar with ICER reports,” says Michelle Simon, PhD, ND, President and CEO of the Institute for Natural Medicine. “Independent third party assessments are extremely valuable in providing a transparent analysis from which improvements can be made. I hope that the learning from this analysis will extend beyond mandating proof of efficacy for fast tracked pharmaceuticals to including natural and non-pharmacologic interventions being recognized and approved for their health enhancing benefits.”
The BMJ report suggests that one effective reform could be that confirmatory trials are designed, agreed, and even started as part of the approval, says Mahase. Other suggestions set out in the ICER white paper include strengthening the selection of surrogate endpoints, regulating the price of accelerated drugs, and regularly re-reviewing and renewing the approval to ensure that it continues to justify the risk benefit tradeoff.
Despite the concerns raised, Mahase points out that all experts who spoke to The BMJ agreed that the accelerated pathway is still useful and can be truly beneficial to patients, although some changes are needed.
In response, an FDA spokesperson said it was “committed to working with sponsors to ensure that confirmatory studies are completed in a timely manner.” They added, “We expect sponsors to commit all resources needed to move trials forward as effectively as possible, with the aim of completing trials as soon as is feasible, while assuring the quality of the data and the robustness of the results.”
Click Here for the Full Text Study